Hemophilia facts
- Hemophilia is one of a group of inherited bleeding disorders that cause abnormal or exaggerated bleeding and poor blood clotting.
- Hemophilia A and B are inherited in an X-linked recessive genetic pattern, so males are commonly affected while females are usually carriers of the disease.
- Hemophilia A is caused by a deficiency of clotting Factor VIII, while hemophilia B (also called Christmas disease) results from a deficiency of Factor IX.
- Hemophilia varies in its severity among affected individuals.
- Symptoms include excessive bleeding from any site in the body; long-term damage to joints from repeated bleeding episodes is characteristic.
- Treatment involves coagulation factor replacement therapy.
- The formation of inhibitors to the treatment factor concentrates is a significant complication of treatment.
- Gene therapy treatments are a source of active research and hold promise for the future.
What is hemophilia?
- Hemophilia is not one disease but rather one of a group of inherited bleeding disorders that cause abnormal or exaggerated bleeding and poor blood clotting. The term is most commonly used to refer to two specific conditions known as hemophilia A and hemophilia B, which will be the main subjects of this article. Hemophilia A and B are distinguished by the specific gene that is mutated (altered to become defective) and codes for a defective clotting factor (protein) in each disease. Rarely, hemophilia C (a deficiency of Factor XI) is encountered, but its effect on clotting is far less pronounced than A or B.
- Hemophilia A and B are inherited in an X-linked recessive genetic pattern and are therefore much more common in males. This pattern of inheritance means that a given gene on the X chromosome expresses itself only when there is no normal gene present. For example, a boy has only one X chromosome, so a boy with hemophilia has the defective gene on his sole X chromosome (and so is said to be hemizygous for hemophilia). Hemophilia is the most common X-linked genetic disease.
- Although it is much rarer, a girl can have hemophilia, but she would have to have the defective gene on both of her X chromosomes or have one hemophilia gene plus a lost or defective copy of the second X chromosome that should be carrying the normal genes. If a girl has one copy of the defective gene on one of her X chromosomes and a normal second X chromosome, she does not have hemophilia but is said to be heterozygous for hemophilia (a carrier). Her male children have a 50% chance of inheriting the one mutated X gene and thus has a 50% chance of inheriting hemophilia from their carrier mother.
- Hemophilia A occurs in about 1 out of every 5000 live male births. Hemoplilia A and B occurs in all racial groups. Hemophilia A is about four times more common than B. B occurs in about 1 out of 20- 30,000 live male births.
- Hemophilia has been called the Royal Disease because Queen Victoria, Queen of England from 1837 to 1901, was a carrier. Her daughters passed the mutated gene on to members of the royal families of Germany, Spain, and Russia. Alexandra, Queen Victoria’s granddaughter, who became Tsarina of Russia in the early 20th century when she married Tsar Nicholas II, was a carrier. Their son, the Tsarevich Alexei, suffered from hemophilia.
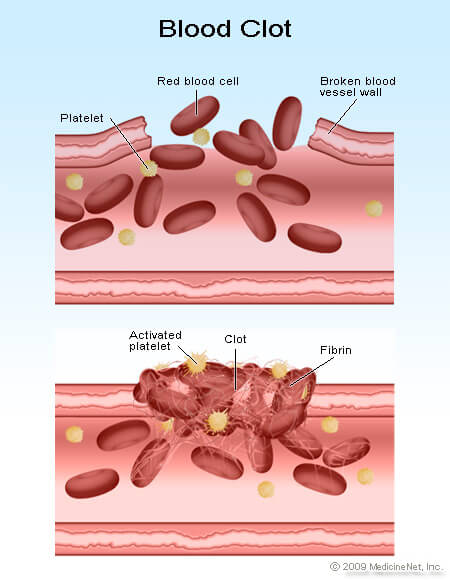
What causes hemophilia?
- As mentioned above, hemophilia is caused by a genetic mutation. The mutations involve genes that code for proteins that are essential in the blood clotting process. The bleeding symptoms arise because blood clotting is impaired.
- The process of blood clotting involves a series of complex mechanisms, usually involving 13 different proteins classically termed factors I through XIII and written with Roman numerals. If the lining of the blood vessels becomes damaged, platelets are recruited to the injured area to form an initial plug. These activated platelets release chemicals that start the clotting cascade, activating a series of 13 proteins known as clotting factors. Ultimately, fibrin is formed, the protein that crosslinks with itself to form a mesh that makes up the final blood clot. The protein involved with hemophilia A is factor VIII (factor 8) and with hemophilia B is factor IX (factor 9).
How is hemophilia diagnosed?
The majority of patients with hemophilia have a known family history of the condition. However, about one-third of cases occur in the absence of a known family history. Most of these cases without a family history arise due to a spontaneous mutation in the affected gene. Other cases may be due to the affected gene being passed through a long line of female carriers.
If there is no known family history of hemophilia, a series of blood tests can identify which part or protein factor of the blood clotting mechanism is defective if an individual has abnormal bleeding episodes.
The platelet (a blood particle essential for the clotting process) count should be measured as well as two indices of blood clotting, the prothrombin time (PT) and activated partial thromboplastin time (aPTT). A normal platelet count, normal PT, and a prolonged aPTT are characteristic of hemophilia A and hemophilia B. Specific tests for the blood clotting factors can then be performed to measure factor VII or factor IX levels and confirm the diagnosis.
Genetic testing to identify and characterize the specific mutations responsible for hemophilia is also available in specialized laboratories.
Is it possible to know if you are a carrier of hemophilia?
Since men with the genetic mutation will have hemophilia, a man who does not have the condition cannot be a carrier of the disease. A woman who has a son with known hemophilia is termed an obligate carrier, and no testing is needed to establish that she is a carrier of hemophilia.
Women whose carrier status is unknown can be evaluated either by testing for the clotting factors or by methods to characterize the mutation in the DNA. The DNA screening methods are generally the most reliable.
Prenatal diagnosis is also possible with DNA-based tests performed on a sample obtained through amniocentesis or chorionic villus sampling. Most individuals are seen and tested by consultants who specialize in genetically linked diseases.
What are treatments for hemophilia?
The mainstay of treatment is replacement of the blood clotting factors. Clotting factor concentrates can be purified from human donor blood or made in the laboratory using methods that do not use donor blood. This type of therapy is known as replacement therapy. Clotting factor replacement therapy is carried out by infusing the clotting factor concentrates into a vein, much like a blood transfusion. This type of therapy can be administered at home with proper instruction and training.
Depending upon the severity of the condition, replacement therapy of the deficient clotting factor may be carried out on an as-needed basis (called demand therapy) or on a regular basis to prevent bleeding episodes (known as prophylactic therapy).
People who have mild cases of hemophilia A are sometimes treated with the drug desmopressin, also known as DDAVP. This drug stimulates release of more clotting factor by the body. It is administered either slowly through the intravenous route (IV) or occasionally, in nasal spray form.
Pain relievers may be prescribed for symptom relief, but pain relievers other than aspirin or non-steroidal anti-inflammatory medications (such as naproxen, ibuprofen) must be used, since these types of drugs further inhibit the blood’s ability to clot. Acetaminophen (Tylenol and others) is often given for pain relief.
Inhibitors
A major complication of treatment is the development of so-called inhibitors to the clotting factors. Inhibitors (antibodies) are produced because the body sees the factor concentrates used to treat patients to reduce or prevent bleeding, as foreign and activates an immune response in the patient to destroy the foreign substances (factor VIII or factor IX).
Inhibitors to factor VIII are the most common and occur in about one-third of those with severe hemophilia A and about 1 out of every 50 people with mild or moderate hemophilia A. They typically develop in childhood in those with severe hemophilia A and later in life in milder cases. Inhibitors destroy both the replacement factor VIII concentrates as well as any factor VIII that is present in the body. This is a serious complication of treatment because the factor concentrates are no longer effective in treating the condition. The action of inhibitors to destroy factor VIII concentrates shows different degrees of severity among individuals and can even vary over time in the same individual.
In about two-thirds of cases, the inhibitors disappear on their own or with treatment known as immune tolerance therapy (ITT) or immune tolerance induction (ITI). In cases of severe hemophilia A with persistence of inhibitors, other factor concentrates, such as activated prothrombin complex concentrate or recombinant factor VIIa, are administered to attempt to help control bleeding.
The development of inhibitors to factor IX is much less common and occurs in about 1% of those with hemophilia B. However, these can cause a very serious allergic reaction when factor IX concentrates are given. Immune tolerance therapy to eliminate inhibitors is less successful than with hemophilia A.
Infections
Blood-borne infections, such as the HIV virus and hepatitis B and C, were a major complication of treatment for hemophilia during the 1980s. These infections were transmitted through the factor concentrates and other blood products that were used to treat hemophilia. The use of large blood donor pools to prepare factor concentrates and the lack of specific tests for infectious agents both contributed to the contamination of blood products used to treat hemophilia. By 1985, about 90% of people with severe hemophilia were infected with the HIV virus, and about half of all people with hemophilia were HIV-positive. Today, improved screening and manufacturing practices including virus removing techniques as well as the development of recombinant factors have essentially eliminated this tragic complication of treatment.
Can hemophilia be prevented?
Hemophilia is a genetic (inherited) disease and cannot be prevented. Genetic counseling, identification of carriers through molecular genetic testing, and prenatal diagnosis are available to help individuals understand their risk of having a child with hemophilia.
What is the outlook (prognosis) for hemophilia?
Before factor concentrates were developed, those with hemophilia had a significantly decreased life expectancy. Life expectancy before the 1960s for those with severe hemophilia was limited to 11 years. Currently, the mortality (death) rate for males with hemophilia is twice that of healthy males. As mentioned before, the increase in HIV and hepatitis infections associated with therapy during the 1980s led to a corresponding increase in death rates.
Currently, prompt and adequate treatment can greatly reduce the risks of life-threatening bleeding episodes and the severity of long-term damage to joints, but joint deterioration remains a chronic complication of hemophilia.
References: